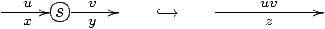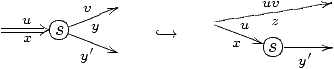Assemblage de fragments d'ADN
Georges Gonthier
georges.gonthier@inria.fr
Dans la course au séquençage de tous les génomes de la biosphère
l'informatique joue un rôle de tout premier plan. En effet, les
procédés physico-chimiques ne permettent de « lire » que des courtes
chaînes d'ADN (typiquement 500 bases), alors qu'un génome en comporte
quelques dizaines de milliers à quelques dizaines de milliards pour
les plus complexes comme le blé (celui de l'homme a trois milliards de
bases). Les biologistes en sont donc réduits à lire une multitude de
petits gragments répartis aléatoirement, puis à tenter
d'assembler les morceaux de ce gigantesque puzzle.
L'informatique s'est révélée un instrument puissant pour cet
assemblage; une compagnie privée a même fait sensation (et scandale)
en parvenant la première, en trois ans, à séquencer le génome humain,
en s'appuyant sur une solution purement informatique. Le consortium
international public, qui oeuvrait depuis dix ans, utilisait lui
une approche plus conservatrice consistant à construire une « carte
physique » du génome : une énorme collection de gros fragments d'ADN
(quelques dizaines de milliers de bases), soigneusement répertoriés et
conservés sous forme de chromosomes artificiels dans des bactéries (ou
BAC). L'assemblage était limité à ces BAC, dont on établissait la
position physique sur le chromosome par d'autres moyens expérimentaux.
La compagnie en question a gagné la course en assemblant directement
des chromosomes entiers; mais elle garde jalousement ses programmes.
Dans ce projet informatique, on vous propose de programmer un nouvel
algorithme [1], qui, bien qu'il ne soit pas encore utilisé
« industriellement », promet d'améliorer significativement
l'efficacité et la fiabilité de l'assemblage.
1 Données du problème
Le génome d'une espèce est, rappelons-le, composé d'un ensemble de
molécules d'ADN, réparties en chromosomes (voire en plasmides pour
certaines bactéries). Ces molécules sont des polymères formés à partir
de seulement quatre molécules élémentaires, qui ne diffèrent que par
la base aminée qu'elles portent --- Adényne, Cytosine, Guanine, ou
Thymine, que l'on abbrège en A, C,G, T. Leur célèbre forme, la
double hélice, est composée de deux longues chaînes (ou brins)
jumelées et complémentaires: chaque A fait face à un C, chaque G
à un T. On peut donc les décrire abstraitement avec un (long) mot
sur ces quatre lettres; c'est le code génétique de l'espèce.
Ce projet traite uniquement de la détermination de ce code; mais
l'interprétation du code est aussi un défi majeur pour la
bioinformatique.
On peut «lire» expérimentalement le code, mais seulement sur de courts
segments (environ 500 bases). En effet la
méthode consiste à
créer par polymérisation des copies des préfixes du codes, colorés
(littéralement!) en fonction de leur dernière lettre. On trie alors
ces préfixes par poids moléculaire (par électrophorèse) pour lire le
code. Malheureusement, la précision du tri et le rendement de la
polymérisation diminuent avec la longueur des préfixes.
La méthode dite de séquençage aléatoire (shotgun sequencing en
anglais) consiste à effectuer une myriade de lectures sur des segments
(appelés inserts) pris au hasard dans le génome. L'ensemble des fragments lus, mis bout à
bout, couvriraient environ 10 fois la longueur du génome. On
tente ensuite d'obtenir algorithmiquement un assemblage de ces
fragments en utilisant leurs chevauchements:
Malheureusement, il faut ensuite faire un long travail de vérification
expérimentale, car la résolution de ce monstrueux puzzle est forcément
partielle, pour trois raisons :
-
Les recouvrements ne sont pas parfaits en raison des erreurs
expérimentales de lecture.
- Statistiquement, il y a forcément des «trous» (gaps) dans
la couverture des lectures, même si leur distribution est uniforme (et
il y a des biais expérimentaux).
- Surtout, les codes génétiques contiennent de nombreuses
répétitions, dont certaines sont très longues; or les recouvrements
avec des segments répétés peuvent donner de fausses indications sur
l'assemblage des fragments (avez-vous déja essayé d'assembler un de ces
puzzle représentant un tas de pièces de monnaie ?).
C'est ce dernier point qui est le plus gênant, car les deux premiers
admettent des solutions statistiques assez satisfaisantes (pour le
second point, on recoupe les parties moins fiables des lectures). En
conséquence, le résultat d'un assemblage n'est pas un code complet,
mais un ensemble plus ou moins réduit de segments, appelés contigs.
Il reste donc ensuite à déterminer expérimentalement l'ordre des contigs,
dans une phase de finition, qui est en général très coûteuse.
Un schéma expérimental astucieux permet d'alléger considérablement le
travail de finition: on sélectionne des inserts assez longs (2 000
à 40 000 bases, dont on séquence les deux extrémités. Cette
technique, qui porte le joli nom de double-barrelled shotgun
sequencing, double statistiquement la longueur de lecture: en effet,
au lieu de fragments isolés, on a maintenant des paires de
fragments séparés par une distance connue. En outre ces paires permettent
d'enjamber (span) les zones dont le séquençage est difficile,
en particulier (et c'est ce qui nous intéresse ici) le début et la fin
de segments répétés. Il faut toutefois noter que les données d'appariement
sont à utilisées avec précaution: elles sont souvent (10 à 20%) erronées
parce que les longs inserts sont fragiles.
2 L'algorithme eulérien
On peut aborder le problème de l'assemblage naïvement, en agglomérant
graduellement les fragments présentant les meilleurs
chevauchements. Avec de bonnes heuristiques, cette approche marche
assez bien, mais elle donne des résultats faux pour les
segments répétés à plusieurs endroits dans le génome. Pour obtenir des
résultats acceptables, il est donc nécessaire d'écarter les fragments
chevauchant le début ou la fin d'un segment répété, ce qui gonfle
artificiellement le nombre de contigs et complique la phase de
finition.
Au fond, l'assemblage naïf revient à chercher un chemin
hamiltonien (qui passe une fois par chaque sommet) dans le
graphe dont les sommets sont les fragments et les arcs sont les
chevauchements entre fragments; or on ne connaît pas d'algorithme non
exponentiel pour ce problème (il est NP-complet).
Dans ce projet on vous propose d'utiliser une approche différente, due
à P. Pevzner et H. Tang [1]. Dans cette approche, on construit
non pas un ensemble de contigs linéaires, mais un automate fini
qui reconnaît toutes les séquences compatibles avec les données
expérimentales. Les cycles dans le graphe de cet automate
correspondront donc aux segments répétés du génome dont la position
exacte n'a pas pu être déterminée. Ceci permet non seulement d'éviter
une fragmentation artificielle des contigs, mais en plus d'utiliser
directement les données d'appariement pour l'assemblage, comme nous
l'expliquerons ci-dessous. Notez toutefois que le graphe de l'automate
peut ne pas être connexe, à cause des gaps de lecture.
L'algorithme proposé se décompose en six phases
-
Tout d'abord, on applique un filtre statistique très strict qui
permet en pratique d'éliminer toutes les erreurs expérimentales des
lectures, en corrigeant ou en écartant toutes les données suspectes.
- Ensuite, on construit le graphe de De Bruijn des fragments,
qui constitue une première approximation de l'automate.
- On raffine et on simplifie le graphe de l'automate, de façon à obtenir
un automate où chaque fragment correspond à un chemin linéaire.
- On utilise les chemins de cet automate pour compléter les données
d'appariement, ce qui donne de nouvelles séquences de lecture, et on
utilise ces nouvelles séquences pour raffiner à nouveau le graphe.
- On utilise les données d'appariement pour relier les différentes
composantes connexes de l'automate (scaffolding).
- On réintègre les données écartées dans la première phase pour tenter
de combler les gaps au maximum.
Pour ce projet informatique, on se limitera aux étapes 2, 3 et 4, qui
sont décrites en plus de détail ci-dessous. Ainsi il ne sera pas nécessaire
de se plonger dans les différentes heuristiques statistiques.
2.1 Le graphe de De Bruijn
On prend, comme première approximation de l'automate, le graphe
de De Bruijn d'ordre k du génome (où k est typiquement pris entre
16 et 20). Il s'agit du graphe dont les sommets sont toutes les
sous-chaînes de longueur k-1 du génome, et dont les arcs
correspondent aux sous-chaînes de longueur k du génome. Plus
précisément, si a u b est une sous-chaîne de longueur k
du génome, alors il y a un arc qui va du sommet a u au sommet
u b, qu'on étiquette par a.
Tout chemin du graphe de De Bruijn qui emprunte tous ses arcs
exactement une fois (un parcours eulérien) donne une chaîne ADN
contenant exactement les mêmes sous-chaînes de longueur k que le
génome. Contrairement à la recherche de parcours hamiltonien, qui
demande un temps exponentiel, la recherche de parcours eulérien est
très aisée. Si on connaît la multiplicité des arcs du graphe
(c'est-à-dire que si une chaîne de longueur k apparaît m fois dans
le génome, on a exactement m exemplaires de l'arc correspondant dans
le graphe), on peut même trouver un parcours eulérien en temps
linéaire, avec un parcours en profondeur (cf. la composition
d'informatique fondamentale 2002).
Si on ne connaît pas les multiplicités, alors on peut recalculer en
temps polynomial les multiplicités minimales qui permettent de
parcourir tout le graphe (c'est le problème dit du facteur
chinois [2]): en effet, ce parcours existe si et seulement si
tous les sommets comportent autant d'arcs entrants que sortants (sauf
les sommets correspondant au début et à la fin du génome). Il suffit
donc de trouver d chemins reliant les sommets ayant trop d'arcs
entrants à ceux ayant trop d'arcs sortant, où d est le
déséquilibre total, ce qui peut se faire en itérant au plus d
fois une variante du calcul des plus courts chemins.
En pratique, on ne peut pas calculer exactement le graphe de De
Bruijn, parce que ceci nécéssiterait de connaître par avance la
totalité génome. On peut cependant en obtenir une excellente
approximation, construite à partir de l'ensemble des chaînes de
longueur k apparaissant dans les fragments, plutôt que dans le
génome. Bien sûr, on n'obtiendra pas les multiplicités; en outre, il y
aura quelques sommets et arcs manquants, à cause des gaps de
lecture. Ainsi, le graphe obtenu ne sera en général pas connexe, et il
aura plusieurs sommets source (sans arcs entrants) et
puits (sans arcs sortant).
Il y a de très grandes chances pour qu'il y ait autant de sources que
de puits, et que chaque source ou puits ne soit relié que par un seul
arc au graphe: en effet il y a peu de chance pour qu'un gap se
produise dans les segments répétés, qui sont au moins deux fois plus
couverts par les fragments. On se placera donc sous cette hypothèse
(que l'on vérifiera); on essayera donc de produire M contigs, comme
autant de chemins reliant les M sources aux M puits.
2.2 Détachements et coupures
On ne peut toutefois pas faire l'assemblage simplement par parcours
eulérien du graphe de De Bruijn. Celui-ci est en effet beaucoup trop
ambigu: il a beaucoup de parcours eulériens, et certains sont très
différents du génome réel. Ceci est du au fait qu'on a fortement
dégradé les données expérimentales lors de la construction du graphe,
en découpant des fragments de 500 bases en fragments de 20 bases ou
moins. Chacun des fragments correspond donc à un chemin de longueur
480 dans le graphe de De Bruijn; on ne recherche pas n'importe quel
parcours eulérien du graphe, mais un parcours sur-eulérien qui
contient chacun de ces chemins.
Il n'y a pas d'algorithme évident pour calculer de tels
parcours. L'idée central de l'algorithme de Pevner et Tang est de
procéder à une série de transformations sur le graphe pour tenter de
ramenr le problème à un parcours eulérien. Si á G0, C0
ñ est la paire initiale ágraphe de De Bruijn, chemins
associés aux fragmentsñ, on obtient une séquence de récritures
qui aboutit (idéalement) à une paire telle que CN soit simplement
l'ensemble des arc de GN. Chaque transformation doit être
correcte, en ce sens que les chaînes correspondant aux parcours
sur-eulériens de á Gi+1, Ci+1ñ doivent être les
mêmes que celles correspondants à celles des parcours sur-eulériens de
á Gi, Ciñ pour tout i<N.
Pour y parvenir il faut étiqueter les arcs des Gi par des chaînes
ADN plutôt que par des bases isolées (d'ailleurs, il fallait déja le
faire pour les arcs menant aux puits de G0). Ainsi, un exemple
élémentaire de transformation correcte consiste à contracter un sommet
s de Gi n'ayant qu'un seul arc entrant x et un seul arc
sortant y, respectivement étiquetés u et v; on élimine s et on
remplace x et y par un nouvel arc z:
Dans Ci, on remplace la séquence xy par z partout où elle
apparaît; on remplace également les y initiaux et les x terminaux
par z: si on note, respectivement, Cixy, Ci®
x, Ciy® les sous-ensembles de Ci qui contiennent
la séquence xy (respectivement se terminent par x, commencent
par y), et que ces ensembles sont disjoints, on obtient Ci+1 en
remplaçant les chemins s x y s'Î Cixy par s z
s', les chemins s xÎ Ci® x par s
z, et les chemins y s' Î Ciy® par z s'
(on effectue des substitutions multiples si Cixy,
Ci® x, Ciy® ont un intersection).
Cette transformation reste valable si x et y ne sont pas les seuls
arc entrants ou sortants de s, mais sont tous deux des arcs
simples. Dans ce cas, on ne supprime pas s, et la
transformation s'appelle le détachement de la séquence xy:
La transformation est correcte dès lors que Cixy n'est pas vide:
en effet puisque x et y ne sont pas multiples, le parcours
sur-eulérien ne passe qu'une seule fois par ces arcs, et puisqu'il
contient un chemin de Cixy, il emprunte y après x. Le
parcours sur-eulérien contient donc un chemin s x Î
Ci® x si et seulement si il contient s xy,
lequel correspond à la même chaîne que s zÎ Ci+1.
En revanche, cette transformation n'est pas correcte si par
exemple x est un arc multiple (donc la fin d'un segment répété). En
effet dans ce cas un chemin s x Î Ci® x
pourrait apparaître suivi d'un arc y'¹ y dans le parcours
sur-eulérien, et il ne serait donc pas correct de remplacer x
par z dans ce cas. Toutefois, si tous les arcs sortants de s sont
simples, on peut tenter de résoudre cette ambiguité en recoupant les chemins:
-
Deux chemins de Ci sont dits compatibles si leur union
est encore un chemin linéaire: une paire de chemins compatibles est
donc soit de la forme {ss', s's''} (chevauchement),
soit de la forme {s', ss's''} (inclusion).
- La séquence xy est détachable si Cixy n'est pas vide
et que tout chemin de
Ci® x est incompatible, soit avec un chemin de Cixy,
soit avec un chemin de Cixy' pour chaque y' tel que Cixy'
ne soit pas vide.
- Dans le détachement de xy, on ne supprime pas x de Gi,
et on ne remplace les x terminaux par z que pour les
chemins de Ci® x qui sont incompatibles avec les Cixy'.
En effet chaque x qui apparaît dans un parcours sur-eulérien est suivi
soit de y, soit d'un des y', et chacun de ceux-ci n'apparaît
qu'une seule fois. Si s x apparaît dans le parcours mais est
incompatible avec un chemin de Cixy' pour chaque y'¹ y,
alors il est forcément suivi de x.
Si c'est y qui est multiple, alors on impose une condition analogue
pour les chemins de Ciy®, et la transformation reste
correcte si x et y sont tous deux multiples, que les deux
conditions sont vérifiées, et que tous les autres arcs sont
simples. Bien qu'il soit facile de trouver des contre exemples
théoriques, en pratique les conditions de détachabilité ne sont pas
vérifiées lorsqu'il y a trop d'arêtes multiples, et donc on peut
éviter de calculer la multiplicité des arcs sur le graphe initial
(au demeurant, il faudrait refaire ces calculs, car si l'heuristique
de multiplicité minimale identifie correctement les arc multiples,
elle peu faire des erreurs sur la valeur exacte de la multiplicité).
En revanche, le détachement n'est pas correct lorsque les conditions
de détachabilité ne sont pas vérifiées. Il se peut toutefois qu'un
détachement initialement impossible le devienne à la suite d'autres
transformations sur le graphe; ainsi, sur des données réelles on
trouve que 95% des séquences d'arcs sont détachables.
Il reste toutefois 5% d'arcs ambigus, qui ne sont détachables ni à
gauche, ni à droite. La situation caractéristique est la suivante
-
Un arc multiple x va de s à s'; x est le seul
arc sortant de s, et le seul arc entrant de s'; par contre s a
plusieurs arcs entrants, et s' plusieurs arcs sortant.
- Cix ® È Ci® x est l'ensemble de
tous les chemins de Ci contenant x: dans Ci, x ne peut être
qu'au début ou à la fin d'un chemin.
Un tel arc x est supprimable. La transformation
correspondante s'appelle une coupure; elle consiste à laisser
G inchangé, mais à supprimer x de tous les chemins de Ci, sauf pour
le chemin de longueur 1 x, que l'on rajoute.
La pratique montre que ces deux transformations, détachements et
coupures, suffisent à ramener le problème du parcours sur-eulérien à
celui du parcours eulérien, en tout cas sur les données expérimentales.
2.3 Paires d'extrémités
La phase suivante de l'algorithme utilise les données d'appariement
pour lever une bonne parite des ambiguités qui persistent dans le graphe
transformé. L'idée est la suivante:
-
Dans le dernier graphe, chaque fragment correspond à un arc, avec
un décalage entier dû aux détachements et coupures.
- Un insert correspond donc à une paire d'arcs (x,y), et une
longueur, qui doit être la longueur d'un chemin commençant en x et
se terminant en y (où la longueur d'un arc est celle de la chaîne
qui l'étiquette).
- Si il y a exactement un chemin de x à y qui a la
bonne longueur, alors ce chemin est le séquencement de l'insert; on
peut donc rajouter un fragment de 2 000 à 10 000 bases aux
données expérimentales!
On peut alors recommencer à faire des détachements et coupures avec
ces nouvelles données, ce qui éliminera encore des ambiguités. Ce
raffinement va éliminer des chemins superflus dans le graphe, ce qui
va permettre d'utiliser d'autres inserts, donc il est avantageux
d'itérer le cycle recherche de chemins/détachements plusieurs fois.
En pratique, il ne subsiste souvent à la fin que des ambiguités dues à
des segments répétés plus longs que les inserts, et donc irréductibles
même en théorie. Soulignons enfin que la méthode s'applique même si la
longueur n'est connue qu'approximativement, et même en présence
d'appariement erronés (puisqu'on ignore les x,y qui ne sont pas
connectés par un chemin de la bonne longueur).
3 Travail demandé
Il s'agit de mettre en oeuvre l'algorithme de Pevner et Tang pour
des lectures simulées, sans erreurs.
-
Votre programme travaillera principalement avec des fichiers.
Il aura en entrée deux fichiers texte, un contenant le génome à
assembler (chaîne de A, C, G, T coupée en lignes), l'autre les
paramètres de l'expérience (nombre et longueurs d'inserts, longueur
des lectures, initialisation du générateur aléatoire, ordre du graphe
de De Bruijn, etc). Votre programme devra pouvoir produire l'automate
final, sous un format que vous définirez. Optionellement,
votre programme pourra écrire et lire l'«expérience» qu'il simule.
- Votre programme devra simuler une expérience de séquencement,
puis appliquer l'algorithme d'assemblage décrit dans la section précédente:
construction du graphe de De Bruijn, détachements, et complétion des inserts.
Il devra ensuite pouvoir comparer ses résultats au vrai génome.
- Bien qu'il ne soit pas nécessaire que votre programme fonctionne
avec un génome complet, vous devrez prendre soin de choisir des
structures de données raisonnablement efficaces. En particulier,
l'utilisation du hachage est indispensable pour la construction du
graphe initial.
- On attend de vous une attitude critique vis à vis de votre travail.
Essayez plusieurs génomes, plusieurs taux de couverture des lectures,
plusieurs ordres de graphe et comparez les résultats obtenus.
Il sera difficile de vous attaquer au problèmes de correction d'erreur dans
le volume horaire du projet. D'autres extensions sont par contre possibles:
-
Vous pouvez calculer les multiplicités dans le graphe final, et
produire les différents ensembles de contigs possibles.
- Vous pouvez utiliser les données d'appariement pour relier les
contigs entre eux et ainsi produire un échafaudage de la
séquence complète.
- Vous pouvez réaliser un affichage graphique des graphes désambigués.
Références
- [1]
- Pavel A. Pevzner amd Haixu Tang, ``Fragment Assembly with Double-Barrelled Data'',
Bioinformatics Discovery Note, 1(1), pp. 1--9, 2001.
http://nbcr.sdsc.edu/euler/euler.htm.
- [2]
- Harold Thimbleby, ``The Directed Chinese Postman Problem'',http://www.uclic.ucl.ac.uk/harold/
Ce document a été traduit de LATEX par
HEVEA.